

17.06.2021
![]()
MSn-Analyse nicht-derivatisierter und Mtpp-derivatisierter Peptide
Dr. Dorota Gaszczyk, Prof. Zbigniew Szewczuk, Dr. Alicja Kluczyk, University of Wroclaw
Ein LC-MS-IT-TOF koppelt die Techniken der Ionenfalle (QIT) und der Flugzeitanalyse (TOF - Time-of-Flight). Die QIT-Ionenfalle bietet leistungsstarke MSn-Fähigkeiten. Nach Auswahl des geeigneten Vorläufer-Ions lassen sich mehrere MSn-Experimente im Verlauf einer einzigen Probenaufgabe durchführen. Es werden Produkt-Ionen erhalten, die als Vorläufer-Ionen für die nächste Fragmentierungsstufe dienen. MSn-Spektren mit einer hohen Massengenauigkeit liefern eine verbesserte Zuverlässigkeit der Signalzuordnung und eine Strukturanalyse durch den Fragmentierungsablauf.
Ein IT-TOF-Gerät bietet hohe Auflösung und Genauigkeit für alle MS- und MSn-Modi ebenso wie Massenstabilität und ausgezeichnete Auflösung für LC-MS-Analysen. Solche Eigenschaften machen ein LC-MS-IT-TOF zum bevorzugten Instrument für moderne wissenschaftliche Untersuchungen zur Verunreinigungsanalyse, für die Erstellung metabolischer Profile und zur Biomarker-Forschung [1].
Die Forschungsprojekte der Arbeitsgruppe für Chemie und Stereochemie von Peptiden und Proteinen der Chemischen Fakultät der Universität Wroclaw, Polen, untersucht Peptide und Proteine hauptsächlich mit Hilfe der Massenspektrometrie. Professor Zbigniew Szewczuk und sein Team erforschen die Struktur natürlicher und synthetischer Peptide, Peptidkonjugate und -addukte, indem sie nach posttranslationalen Modifikationen und Biomarkern suchen. Auf Grund der geringen Konzentration einiger Peptide in biologischen Proben besteht in der Empfindlichkeitssteigerung der MS-Analyse eines der wichtigsten Forschungsziele der Arbeitsgruppe.
Zwei Beispiele für aktuelle Untersuchungen werden hier vorgestellt, die aufgrund der benötigten MSn-Analytik nur durch den Einsatz eines LC-MS-IT-TOF-Systems möglich wurde.
1. MSn-Analyse mit 4-(4-Methoxyphenyl)-2,6-diphenylpyryliumsalzderivatisierter Peptide mit LC-MS-IT-TOF
Tandemmassenspektrometrie ist ein leistungsfähiges Werkzeug in der Proteomikforschung. Trotz der rapiden Entwicklung dieser Technik besteht noch immer das Problem unzureichender Empfindlichkeit wegen der schlechten Ionisierung einiger Peptide, die in der biologischen Probe in geringer Konzentration vorhanden sind. Der Einbau einer funktionellen Gruppe erlaubt, dieses Problem zu umgehen. Sie bringt eine stabile positive Ladung innerhalb des Peptidmoleküls ein, steigert damit die Ionisierungseffizienz und ermöglicht einen empfindlichen Nachweis mit der Elektrospray-Massenspektrometrie (ESI-MS).
Kürzlich haben Forscher diverse effiziente Methoden zur Peptid-Derivatisierung durch eine quartäre Ammoniumsalz(QAS)-Synthese entwickelt [2]. Die höchste Intensitätssteigerung (etwa 1.000-fach) wurde bei Pyrylium-Ionisierungsreagenzien beobachtet, die leicht mit der primären Aminogruppe in der Lysinseitenkette reagieren, um Pyridiniumsalze mit stabiler positiver Ladung zu bilden [3]. Die mit Pyrylium-Reagenzien derivatisierten Peptide lassen sich sogar auf attomolarem Niveau (10-18) nachweisen. Eine neue Art von isobarer Peptid-Markierung wurde ebenfalls vorgeschlagen [4], die auf dem Austausch der 16O/18O-Paarung und der Derivatisierung von Isotopologen der Pyridiniumsalze beruht.
Allerdings ist es notwendig, nach neuen Ionisationsmarkern zu suchen, die einen vergleichbaren Grad an Ionisierungssteigerung zeigen und verbesserte Eigenschaften besitzen (höhere Löslichkeit, Empfindlichkeit und Analysepräzision). Forscher entwickelten kürzlich ein neues Ionisierungsreagenz (4-[4-Methoxyphenyl]-2,6-diphenylpyryliumsalz, Mdppl), um die Anwendbarkeit in der Proteomikforschung zu verbessern (unveröffentlichte Daten). Spezifische MSn-Übergänge werden für die eindeutige Analyse bestimmter, mit Mdppl derivatisierter Peptide eingesetzt.
Das 4-(4-Methoxyphenyl)-2,6-diphenylpyryliumsalz wurde mit der Seitenkettenaminogruppe eines Modell-Peptids verknüpft (Lys-Pro-Pro-Pro). Abbildung 1 zeigt die ESI-MS- und ESI-MSn-Analyse von Lys(Mtpp)-Pro-Pro-Pro.
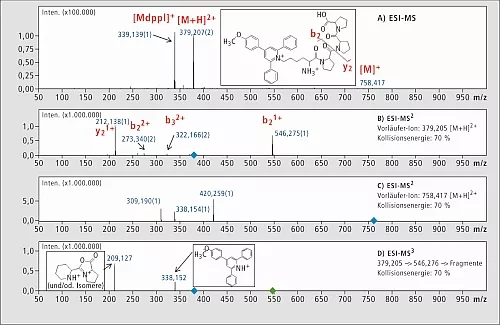
Abb.1: ESI-MS-Spektrum des rohen Synthetik-Peptids Lys(Mdpp)-Pro-Pro-Pro (Tafel A),
ESI-MS2-Fragmentierungs-Spektrum von [M+H]2+ (Tafel B)
und das Fragmentierungs-Spektrum von [M]1+ (Tafel C)
und die ESI-MS3-Analyse von [M+H] 2+ → b2+ → (Tafel D).
(Mdpp - 4-(4-Methoxyphenyl)-2,6-diphenylpyryliumsalz).
Bei MS2-Experimenten wurde eine Reihe von b- und y-Ionen erhalten. Aufgrund des Anstiegs der Kollisionsenergie vergrößerte sich die Stärke des Signals m/z 338, obwohl sie weiterhin recht gering war. Nach dem MS3-Experiment entsprach der Peak bei m/z 209 dem inneren Fragment der Peptidsequenz. Das Signal ist für Pyridiniumsalz charakteristisch, erscheint aber hauptsächlich beim ladungsfernen Fragmentierungsmechanismus [5].
Um diese Aussage eindeutig zu bestätigen, wurde ein MS3-Experiment durchgeführt, bei dem für die erste Fragmentierungsreaktion ein [M+H]2+ ausgewählt wurde, das ein einziges mobiles, für einen ladungsgerichteten Fragmentierungsmechanismus verantwortliches Proton enthält. Resultierende einfach geladene Fragmente enthalten kein mobiles Proton und können einer Fragmentierung gemäß ladungsfernem Fragmentierungsmechanismus unterliegen. Eine Kombination dieser zwei aufeinander folgenden Fragmentierungsreaktionen, die nach verschiedenen Mechanismen ablaufen, können nur mit Hilfe von mit Ionenfallen (IT = ion trap) ausgestatteten Geräten durchgeführt werden.
Eine präzise Analyse der sich ergebenden Fragmente benötigt hochauflösende Spektren, die sich nur mit IT-TOF-Geräten garantieren lassen. Die Forscher stellten fest, dass eine Fragmentierung von einfach geladenen Ionen Informationen über die Ionisierungsstelle gibt, wohingegen das gleiche Experiment für ein zweifach geladenes Ion mehr Information über die Peptidsequenz liefert. In Kombination liefern diese Experimente zuverlässigere und umfangreichere Strukturinformationen. Das Ionenfragment m/z 338 lässt sich beim MRM oder Vorläufer-Ionenscan einsetzen, um all die Peptide zu untersuchen, die 4-(4-Methoxyphenyl)-2,6-diphenylpyrylium-Gruppen enthalten.
Als ein MS3-Ergebnis erscheinen [M+H]2+ → b2+ → Fragmente bei m/z 209 und 338, entweder als [M+H] ]2+ oder [M] ]+ (Abbildung 1B, 1C). Die MS3-Technik führt daher zu eindeutigen Fragmenten, was bedeutet, dass sie sich einsetzen lässt für eine empfindliche und eindeutige Analyse von Anzeige-Ionen bei proteomischen Untersuchungen von mit Mtppl derivatisierten Verbindungen wie beispielsweise: 379 → 546 → 338 und 379 → 546 → 209. Dies eröffnet vollständig neue Möglichkeiten für den Einsatz einer MSn-Analyse bei der quantitativen und qualitativen Proteomik.
Zusammenfassend bleibt festzuhalten, dass sich die MSn-Technik erfolgreich für eine empfindliche Peptidsequenzierung mit hohen Kollisionsenergien einsetzen lässt. Diese schnelle Bestätigung der Struktur derivatisierter Peptide ist extrem zuverlässig. Eine MSn-Analyse ermöglicht die eindeutige Identifikation von Peptidfragmenten, die sich aus MS2-Experimenten ergeben. Die Genauigkeit des IT-TOF-Instruments erlaubt die Summenformel von Fragmenten zu bestimmen, was es möglich macht, Strukturformeln anhand der erhaltenen Ionen vorzuschlagen. Weiterhin bieten sie zudem auch neue und schnelle Methoden zum Nachweis von Biomarkerspuren in den getesteten Proben. MSn-Experimente kombinieren zwei Fragmentierungsmechanismen: ladungsfern und ladungsgerichtet.
2. Innere Fragmente, die im MSn-Verlauf zur Analyse von Peptid-Isomeren gebildet werden
Die Bedeutung der massenspektrometrischen Analyse bei der Peptidforschung ist auf diversen Ebenen offensichtlich - von der Charakterisierung natürlicher oder synthetischer Produkte, über die Sequenzanalytik, bis hin zu Stabilitätsstudien und Quantifizierung. Es gibt auch ein steigendes Interesse an massenspektrometrischen Anwendungen für physikochemische Untersuchungen. Dazu gehören Ladungsvarianten, molekulare Interaktionen und Stabilitätsstudien von zusammengesetzten Molekülen.
Die wesentlichen Vorteile der Massenspektrometrie bestehen in der hohen Empfindlichkeit und der eindeutigen Produktidentifikation mit ausgezeichneten Nachweisgrenzen sowie der Möglichkeit zur Anwendung bei isotopisch modifizierten Referenzmaterialien.
Die Lipophilizität chemischer Verbindungen, ausgedrückt als Logarithmus des Oktanol-Wasser-Verteilungskoeffizienten (logP), ist ein sehr wichtiger Parameter in der medizinischen Chemie. Üblicherweise wird logP mit der traditionellen Flaschen-Schüttelmethode (shake-flask method = SFM) ermittelt, die sehr zeitaufwendig ist, beträchtliche Materialmengen benötigt und im Falle von stark hydrophoben Verbindungen nur begrenzt einsetzbar ist. Zahlreiche chromatographische Methoden wie RP-TLC und RP-HPLC ergänzen oder ersetzen eine SFM. Sie bieten verschiedene praktische Vorzüge, darunter Reproduzierbarkeit, Unempfindlichkeit gegenüber Verunreinigungen und Abbauprodukten, einen größeren Dynamikbereich sowie eine verminderte Probenbehandlung und einen geringeren Probenumfang [6]. Einer der Hauptvorzüge der HPLC bei der Lipophilizitätsbestimmung besteht in der Fähigkeit, unmittelbar die Eigenschaften einer Reihe von Verbindungen durch Analyse von Mischungen zu vergleichen.
Wegen der Blut-Hirn-Schranken-Problematiken wird Lipophilizität intensiv bei der Konzeptionierung und Entwicklung von Neuropharmaka untersucht. Diverse Forschungsgruppen studieren die antinozizeptive Aktivität von Peptiden [7, 8], und es besteht ein erhebliches Interesse an der Anwendung von RP-HPLC zum Vergleich der Lipophilizität der vorgeschlagenen Analoga.
Ein LC-MS-Verfahren erscheint ideal für diese Anwendung, da zusätzliche nützliche Hinweise über den Ladungszustand der untersuchten Peptide erhalten werden. Ein komplexerer Tandem-MS2-Ansatz wird im Falle von isomeren Peptiden benötigt, um die Peak-Reihenfolge den richtigen Probenkomponenten zuzuordnen. Im aktuellen Projekt stießen die Forscher auf ein Problem, das einen fortschrittlicheren Ansatz zur Unterscheidung von Peptidisomeren erfordert. Um den Effekt einer Sequenzmodifikation auf das chromatographische Verhalten von Peptiden und nachfolgend ihre Lipophilizität zu untersuchen, wurde eine Peptidbibliothek konzipiert und aufgebaut, die auf dem Podocin-Fragment 290 - 296 mit der Sequenz Ser-Ile-Ala-Gln-Asp-Ala-Lys basiert. Alanin-Reste wurden als variable Stellen ausgewählt und ersetzt durch Tyrosin, Threonin, Prolin, Glycin und Asparagin, um ein breites Spektrum an Änderungen zur Verfügung zu stellen.
Teilbibliothek 1:
Ser-Ile-Xxx-Gln-Asp-Ala-Lys
Teilbibliothek 2:
Ser-Ile-Ala-Gln-Asp-Xxx-Lys
Wobei Xxx für Gly, Tyr, Thr, Pro, Asn oder Ala steht
Die zwei resultierenden Teilbibliotheken (modifiziert an Position 3 bzw. 6) wurden mit einem LC-MS analysiert. Die Peptide in jeder Teilmenge unterscheiden sich im Molekulargewicht, und die MS-Analyse reichte aus, die Signale zuzuordnen. Über extrahierte Chromatogramme (XIC) dargestellte Elutionsprofile sind in Abbildung 2 gezeigt. Retentionszeit-Unterschiede zwischen Peptidisomeren sind minimal, und der Positionseffekt der modifizierten Rückstände auf die Retentionszeit konnte erst untersucht werden, nachdem eine Methode zur Unterscheidung von Peptid-Isomeren für eine Reihenanalyse erarbeitet werden konnte.
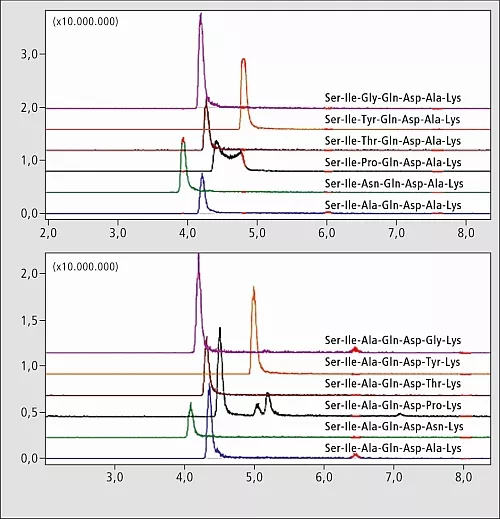
Abb.2: LC-MS-Chromatogramme der Peptid-Teilbibliotheken.
Extrahierte Ionenchromatogramme (XIC) für +2 Ionen abbildende Bibliothekskomponenten.
Die Forscher benutzten die Teilbibliotheken zur MS2-Analyse, indem sie [M+2H]+2-Peptid-Ionen als Vorläufer (Abbildung 3) einsetzten. Die hohe Massenstabilität von TOF erlaubte die Durchführung einer langen Reihe von LC-MS-Experimenten ohne Nachkalibrierung. Vorläufer-Ionen von überlappenden chromatographischen Peaks wurden erfolgreich ausgelesen und aufgrund der hohen Auflösung und Genauigkeit der IT-TOF-Anordnung fragmentiert. Praktisch in allen Fällen erzeugte die Fragmentierung b2-und y5-Ionen, die bei den Peptid-Isomeren paarweise identisch sind und nicht verwendet werden können, um Peaks im Chromatogramm einer Bibliotheksmischung zuzuordnen. Einige beobachtete Unterschiede wurden als +2 Fragment-Ionen (Thr- und Pro-Peptide) identifiziert oder waren ziemlich schwach (Gly-Analog).

Abb.3: MS2-Analyse der Peptid-Teilbibliotheken, [M+2H] 2+-Ionen wurden als Vorläufer ausgewählt
Die Analyse wurde mit einem IT-TOF-Gerät von Shimadzu durchgeführt, und es wurde entschieden, das MSn-Merkmal zu nutzen sowie y5-Ionen der weiteren Fragmentierung (MS3) zu unterwerfen. Die Tyr-Analoga wurden für Voruntersuchungen ausgewählt, und die in Abbildung 4 gezeigten Ergebnisse offenbaren interessante Unterschiede in den MS3-Fragmentierungsmustern. Der charakteristische MS3-Übergang wird in weiteren Lipophilizitätsstudien der Podocin-Peptidfragmentbibliotheken genutzt werden.
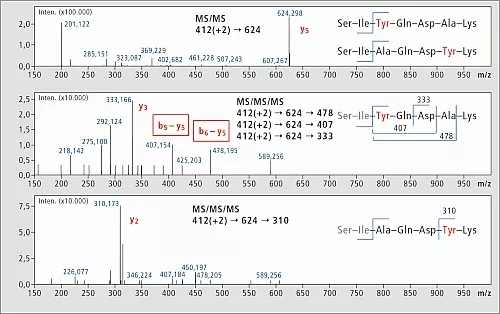
Abb.4: MSn-Analyse von Tyr-enthaltenen Bibliothekselementen.
MS2-Ergebnisse waren nicht ausreichend, charakteristische Ionen zu erzeugen,
wohingegen MS3-Experimente verwendet werden konnten, Isomere eindeutig zu identifizieren.
Basierend auf individuellen Eigenschaften der untersuchten Peptidsequenz konnten normale MS2-Experimente nicht bestätigen, welcher Ala-Rest in einem vorliegenden Peptid ersetzt wurde. Die Peptid-Isomere konnten nur identifiziert werden, nachdem ein tieferer Einblick in die Fragmentierung mit Hilfe der MS3-Funktion der IT-TOF-Anordnung erhalten wurde.
Fazit
Wissenschaftliche Herausforderungen konnten schnell, erfolgreich und effizient gelöst werden dank der MSn-Fähigkeit und der hohen Massengenauigkeit des eingesetzten LC-MS-IT-TOF-Instruments.
Methoden:
Pyryliumsalz-Synthese: Die Synthese von Pyryliumsalzen umfasst zwei Schritte. Der erste besteht in der Synthese eines spezifischen Chalkon, das auf einer Kondensationsreaktion mit verschiedenen, kommerziell verfügbaren Aldehyden und Ketonen beruht. Die Reaktion wird in alkalischer Umgebung durchgeführt - in Gegenwart von 10% NaOH in Ethanol. Im zweiten Schritt wird eine Zyklisierungsreaktion mit dem erhaltenen Chalkon und einem geeigneten Keton angeschlossen. Die Reaktion benötigt das Vorhandensein von Trifluormethanesulfonsäure (TFMSA) als Gegen-Ion.
Derivatisierungsverfahren:
Eine Modellpeptidprobe wurde in Dimethylformamid (DMF) gelöst und ein geeignetes Pyryliumsalz und Trimethylamin (TEA) im Überschuss hinzugefügt. Die Mischung wurde bei 60°C für drei Stunden inkubiert. Nach dieser Zeit wurde das Lösungsmittel unter Stickstoff verdampft und der Rückstand lyophilisiert.
Die Peptid-Bibliothekssynthese wurde mit massiver Unterstützung unter Einsatz von Fmoc-Chemie umgesetzt, und äquimolare Mischungen von Peptid-Derivaten wurden bei verschiedenen Schritten genutzt. Für die LC-MS-Experimente wurden Rohprodukte eingesetzt.
MS- und MSn-Experimente: MS und MSn wurden mit einem IT-TOF-Massenspektrometer von Shimadzu vorgenommen. Für Experimente im Vorläufer-Ionenmodus wurde ein dem Pyryliumsalz entsprechendes Fragment-Ion ausgewählt.
LC-MS-Bedingungen: Eine UHPLC Nexera, ausgestattet mit einer Aeris Peptide XB-C18-Säule (50 x 2,1 mm, 3,6 μm) wurde eingesetzt unter Verwendung einer Gradientenauftrennung von 5 bis 60% B in 15 min, A: 0,1 % HCOOH in Wasser, B: 0,1 % HCOOH in Acetonitril, Flussrate 0,2 ml/min. Das IT-TOF-Gerät wurde im positiven Ionen-Modus betrieben, und der 150 - 1.000 m/z-Bereich wurde analysiert.
Danksagungen
Die Forschung wurde teilweise unterstützt durch die Forschungsförderung Nr. UMO-2016/23/B/ST4/01036 vom Nationalen Wissenschaftszentrum in Polen. Die Autoren danken O. Uysal und P.U. Godigamuwa Acharige für die Bibliothek-Synthese. Ein Dank geht auch an Andrzej Reszka (Shim-Pol, Polen) für den Zugang zum IT-TOF-Instrument von Shimadzu.
Dieser Artikel wurde zuerst in den Shimadzu NEWS 2-2020 veröffentlicht.
Literatur
- Liquid Chromatograph-Mass Spectrometry
- M. Cydzik, M. Rudowska, P. Stefanowicz, Z. Szewczuk, J. Pept. Sci. 17 (2011), 445-453
- M. Waliczek, M. Kijewska, M. Rudowska, B. Setner, P. Stefanowicz, Z. Szewczuk, Sci. Rep., 6 (2016), 1-12
- M. Waliczek, R. Bachor, M. Kijewska, D. Gąszczyk, K. Panek-Laszczynska, A. Konieczny, K. Dabrowska, W. Witkiewicz, K. Marek-Bukowiec, J. Tracz, M. Luczak, Z. Szewczuk, P. Stefanowicz, Anal. Chimica Acta, 1048 (2019), 96-104
- M. Rudowska, D. Wojewska, A. Kluczyk, R. Bachor, P. Stefanowicz, Z. Szewczuk, J. Am. Soc. Mass Spectrom., 23 (2012), 1024-1028.
- C. Liang, J. Qiao, H. Lian, J. Chrom. A, 1528 (2017) 25-34
- H. Liu, B. Zhang, X. Liu, Ch. Wang, J. Nib, R. Wanga, Bioorg. Med. Chem. 15 (2007) 1694-1702
- K. Wtorek, R. Artali, J. Piekielna-Ciesielska, J. Koszuk, A. Kluczyk, L. Gentilucci, A. Janecka, Eur. J. Med. Chem. 179 (2019) 527-536